Research Summary
My research interests cover quite a wide area of high performance and scientific computing. First, I focus on code parallelization and performance optimization (from hand-tunning of existing applications to automatic methods improving code efficiency). Second, I am interested in many areas of applied computing, such as computational problems from chemistry and biology, or real-time modelling of deformations (e.g. simulations of soft tissues).
I focus on code optimization and parallelization (mainly single-node parallelism). Namely, I develop a source-to-source compiler allowing CUDA kernel fusions and Kernel Tuning Toolkit, which can be used for application-level autotuning of OpenCL or CUDA code. I am also interested in application of architecture-aware programming (i.e. optimizing existing codes) for x86CPUs, MICs and GPUs.
In area of scientific computing, I develop a CaverDock software, which enables to compute trajectory and energy profile of a ligand passing through protein's cavity. I also collaborate with CNB CSIC on acceleration of Xmipp (software for image reconstruction in cryo-electron microscopy). I have also participated on development of experimental systems for real-time interactive simulations of molecular docking and soft tissues deformations.
Interests
- Methods for Automatic Code Optimization
- Runtime Systems
- Performance Engineering
- Performance Prediction Methods
- Computational Chemistry and Biology
- Real-time Simmulations
Active Research Projects
-

The OpenCL is open standard allowing to program many types of accelerators as well as classic CPUs. It allows to implement highly parallel computational kernels executed on computing devices. The development and optimization of kernels is challenging even for experienced programmers, as it requires to efficiently parallelize the code to thousands of independent thread and follow many performance characteristics of current hardware (which changes significantly for different hardware types and even generations). Thus, any automatic tool, which eases the exploration of performance-related parameters, has a great value in this area.
We develop Kernel Tuning Toolkit (KTT). With KTT, a programmer identifies parameters of the code, which may have influence on the performance, and the KTT automatically searches the parameter space and picks configurations which leads to the highest performance on particular hardware device. The autotuning decreases the time needed for manual exploration of code tuning parameters and allows developers to write flexible codes, which optimize themselves for underlaying hardware architecture automatically. KTT extends state-of-the-art autotuning methods by allowing to tune kernels compositions with possible host overhead and allows better control of numerical precision. It has also ability to tune kernels dynamically, i.e. during application runtime. We are currently working towards designing new tuning search space methods.
-

The understanding of biological activity of proteins is crucial in many disciplines, such as drug design or enzyme engineering. The important interaction of protein and small ligand molecule is performed in protein's active site. The active site may be hidden in the protein cavity, and thus the transport of ligand into active site needs to be studied. Current computational methods for transport process study may be divided into two main categories -- the geometrical methods and methods based on molecular dynamics. The geometrical methods are very fast and easy to setup, but they consider ligand and protein as macro-world bodies, without utilization of chemical forces. Thus, they can be used for quantitative analysis only. On the contrary, methods based on molecular dynamics simulate the protein-ligand interaction using chemical force field. They results in molecules trajectory (i.e. atoms movement) and energy. However, those methods are computationally demanding and hard to setup.
We are developing a novel method and tool for transport process analysis. Our method uses modified molecular docking based on Autodock Vina, which docks ligand using constraints allowing to precisely restrict the position in the protein cavity. By iterative docking the ligand along the protein cavity, we obtain its trajectory and also energy profile. The heuristic algorithm is used to determine the constraint for molecular docking, thus much less protein-ligand conformations are evaluated compared to molecular dynamics resulting to faster simulation. Our method is implemented in CaverDock tool, which uses geometrical tunnels from well-known software Caver and docks into the tunnel by using our modification of Autodock Vina. The CaverDock is very easy to setup and needs significantly less computational time comparing to methods based on molecular dynamics, whereas it is still able to return trajectory of ligand movement and energetic profile of the transport process. Currently, we are working towards improving heuristics and adding more accurate receptor's flexibility into CaverDock.
-
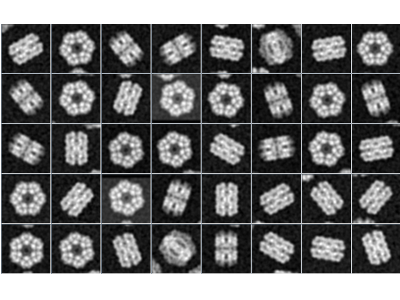
The cryo-electron microscopy (cryo-EM) is very quickly evolving method with growing popularity in structural biology. It allows to study specimen in its natural environment (e.g. no crystalization is needed), thus is highly relevant in many applications. The progress in electron detectors and image processing in recent few years allows cryo-EM to image specimens on nearly atomic resolution. However, the post-processing of images gathered from electron microscopes is highly computationally demanding, as (i) the noise-to-signal ratio is very poor, (ii) the 3D specimen needs to be reconstructed from 2D images of unknown view position and (iii) there may be several conformations of specimen observed.
One of very popular tool for processing cryo-EM data is Xmipp, developed by Biocomputing Unit, Spanish National Center for Biotechnology. As the image reconstruction is highly computationally demanding, we focus to speed-up the software to use computing facility more efficiently. Currently, we have implemented a vectorized version of image alignment in Xmipp, improving its speed more than 2x on classic CPUs. Moreover, we have implemented a GPU version of 3D Fourier reconstruction and movie alignment, both bringing an order of magninude speedup. In the future, we plan to accelerate a code for 3D classification.
-
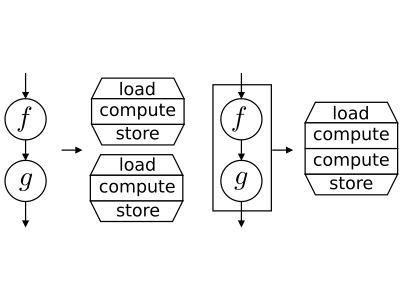
The OpenCL Kernels can be executed on a broad range of hardware devices, providing various level of concurrency, using different caching mechanism and introducing various kernel execution overhead. Thus, searching for optimal granularity of computational kernels is not an easy task. Larger kernels may introduce lower overhead, higher parallelism, and better memory locality, but may also consume more resources such as registers or cache and thus be inefficient.
In this research, we are focused to a kernel fusion method. When the computation is realized by multiple kernels, their fusion may improve data locality, parallelism or serial efficiency. However, it is highly impractical to develop libraries of fused kernels as the number of potential combination of kernels is very high and fusion may decrease performance in some cases. Instead, the programmer may define the computation as a data flow between simple kernels and the source-to-source compiler creates fusion automatically according to data dependencies between kernels and targeted hardware device. We have developed a source-to-source compiler performing fusion on kernels performing (potentially nested) map and reduce operations. Currently, we are extending kernel fusion to more generic kernels.